Genome-speak left the message incomplete: genes lead to proteins, yes, but there’s something more beyond translation. You need the protein sequence.
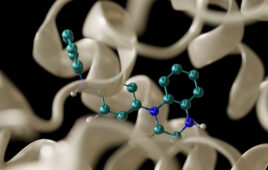
 click to enlarge Shotgun protein sequencing: Assembling MS/MS spectra from overlapping peptides into protein sequences. (Source: Nuno Bandeira, University of California, San Diego) |
In the beginning, there was Edman degradation—the lopping off N-terminal residues, the sum of which revealed a protein’s hitherto unknown sequence. And it was good. Yet, “The goal of my research was to beat out the Edman sequencer,” says Don Hunt, professor of chemistry and pathology, University of Virginia, Charlottesville. “We finally beat them in sensitivity around 1983 and when I did that, Lee Hood (a pioneer of protein sequencing) asked if I would send him one of my students.” Hunt sent John Yates, who is now one of the top researchers in the proteomics field. Yates arrival heralded the departure of the once powerful Edman, and the age of the mass spectrometer came to pass.
The advent of electrospray ionization allowed the marriage of the HPLC separation column to the mass spec. Then people started digesting peptides, clipping them into smaller, more manageable bits and firing them at sophisticated analyzers. “That’s called shotgun proteomics,” says Hunt. “Yates is one of the pioneers for that, and that’s what everybody’s been using since the 1990s. But the problem with that technology is that it ignores all the post-translational modifications— what really makes a protein important.” Proteins identified through mass spec are not usually sequenced, per se, but spectra from a few peptide fragments are mapped back to a known genome, and the sequence is then read from the base pairs. But post-translational modifications involve regulation—important stuff. Proteins also often exist as splice variants—not something the DNA can reveal. “Shotgun proteomics doesn’t give you any idea which splice variant you’re dealing with,” Hunt says.
|
The next innovation, by McLafferty et al., (Science. 2006) involved the use of near-thermal energy electrons, which allowed analysis of intact proteins, including modifications. Hunt considered this exercise a solid proof of principle, but not analytically practical. He prefers the method recently developed in his lab called electron transfer dissociation (ETD) mass spec. The advantage is one of time. “The reaction is very efficient, happening in milliseconds, not hours,” says Hunt. “And you don’t have to buy a new mass spec, it works on all the commercial instruments now used.” The technique can identify and sequence splice variants, as well as shepherd through to analysis those post-translational modifications, which are otherwise quite labile.
Sugar, sugar, snake
Sugar. One such post-translational modification is tacking on something sweet. “The overriding goal in my lab is to characterize glycosylation of proteins,” says Ron Orlando, PhD, assistant professor of chemistry, University of Georgia, Athens. “That sounds like there should be a whole bunch of people doing that—there aren’t.” It’s not an easy thing to do. First, we’re often talking about modifications on membrane proteins—the problem child of proteomics. Second, glycans are a highly-variable modification, consisting of a few to many sugar subunits, in conformations (linear or branched), which are, worst of all, sensitive under interrogation. “They tend to fall apart. A lot of people who work with proteins ignore the carbohydrate part,” says Orlando. “Other people just cut all the carbohydrates off and see what they are …” But information is lost. Glycoslylation is known to change when cells turn cancerous (think, biomarker). Further, alterations occur during cell differentiation, which is of particular interest to people doing stem cell research. “Stem cell people want markers so that they can better purify out certain populations. Tumor people typically are looking at antibody-targeted, drug delivery-type systems—if you can find a unique protein on the surface,” Orlando says. Currently, he is using a tandem mass spec configuration, and the ProteoIQ software he helped develop for the proteomics company, Bioinquire, Athens, Ga., to solve his protein puzzles. [Orlando is on the scientific board of Bioinquire.]
Snake. What do you do if there is no known genome standard to search a peptide mass/charge ratio against, like, say, from a snake? Nuno Bandeira, executive director of the Center for Computational Mass Spectrometry, University of California, San Diego, encountered just such a problem while working with snake venom. “Most likely we [purchased] venom from multiple snakes, not just one. And we indeed found a number of sequence variants, some known, some not,” he explains. To untangle this toxic mixture, Bandeira developed software that is able to both sequence proteins de novo, and discriminate between the subtle shifts in mass that give character to splice variants. The mass spec hardware remains the same—the difference was the innovation of interpreting overlapping peptide spectra. “It’s essentially computational processing to find these spectra and deciphering the ensemble that results. Eventually that builds a ladder that spans large portions of the protein.”
Being able to identify variation is the order of the day. “A prime example would be immunoglobulins, antibodies that are created by recombination—new sequences are created all the time,” Bandeira says. And biomarkers. “Especially in those cases where the biomarker was a different modification of some sort,” says Bandeira, “because that creates these pairs that alignment (software) capitalizes on. It doesn’t need to be told which modifications to look for so it would essentially try any possible mass difference between the two peptides and let the data decide which modifications agree with the spectra.”
Outside interest in his work has been keen, including collaboration with Genentech, South San Francisco, Calif., for sequencing antibodies, and inquiries from Amgen, Thousand Oaks, Calif., about tweaking the search engine’s code.
The software is open source, and currently available to any interested party. For further information see: https://peptide.ucsd.edu/Software/SpectralNetworks.html.
About the Author
Neil Canavan is a freelance journalist of science and medicine based in New York.
This article was published in Drug Discovery & Development magazine: Vol. 11, No. 7, July, 2008, pp. 24-29.
Filed Under: Genomics/Proteomics